I tremendously enjoy the television program “How it’s made”. And even though it is unlikely I’ll ever fabricate a baseball bat, at least I’ve learned how it’s done. This post is intended to similarly transfer that sense of wonder, but not about sports equipment, but about the analysis of the veritable code of life: DNA.
Back in 2002, I became fascinated by DNA, and wrote an article called “DNA as seen through the eyes of a computer programmer”. It can be found at http://ds9a.nl/amazing-dna.
Recently, I’ve been able to turn my theoretical enthusiasm into practical work, and as of November, I’ve been working as a guest researcher at the Beaumontlab of the department of Bionanoscience at Delft University.
Arriving there, I was overwhelmed by the sheer amount of file formats and stuff involved in doing actual DNA processing. So overwhelmed that I found it easier to write my own tools than get to grips with the zoo of technologies out there (some excellent, some “research grade”).
This post recounts my travels, and might perhaps help other folks entering the field afresh. Here goes. It is recommended to read http://ds9a.nl/amazing-dna as an introduction first. If you haven’t, please know that DNA is digital, and can be seen as stored in files called chromosomes. Bacteria and many other organisms have only one chromosome, but people for example have 2*23.
A typical bacterial genome is around 5 million DNA characters (or ‘bases’ or ‘nucleotides’), the human genome is approximately 3 billion bases long.
FASTA Files, Reference Genome
We store DNA on disk as FASTA files (generally), and we can find reference genomes at http://ftp.ncbi.nlm.nih.gov/genomes/. For reasons I don’t understand, the FASTA file(s) have a .fna file extension.
When we attempt to read the DNA of an organism, we call this ‘sequencing’. It would be grand if we could just read each chromosome linearly, and end up with the organism’s genome, but we can’t.
While lowly cells are able to perform this stunt with ease (and in minutes or a few hours), we need expensive ($100k+, up to millions) equipment which only delivers small ‘reads’ of DNA, and in no particular order either. Whereas a cell can copy its genome in minutes or hours, as of 2013 a typical sequencing run takes many hours or days.
Sequencing DNA can be compared to making lots of copies of a book, and then shredding them all. Next, a sequencer reads all the individual snippets of paper and stores them on disk. Since the shreds are likely to overlap a lot, it is possible to reorder them into the original book.
There are different sequencing technologies, some deliver short reads of high quality, other longer reads of lesser qualities, and older technologies deliver long reads of very high quality, but at tremendous expense. The current (2013) leaders of the field are Illumina, Life Technologies (Ion Torrent) and Pacific Biosciences. Illumina dominates the scene. The Wikipedia offers this useful table.

Illumina MiSeq, 2013
FASTQ Files, DNA reads
There are various proprietary file formats, but the “standard” output is called FASTQ. Each DNA ‘read’ consists of four lines of ASCII text, the first of which has metadata on the read (machine serial number, etc, mostly highly redundant and repetitive). The second line.. is a plus character. Don’t ask me why. The third line are actual DNA nucleotides (‘ACGTGGCAGC..’), and the final line delivers the Q in FASTQ: quality scores.

This is where the FASTQ comes out (there’s also ethernet)
Error rates vary between reads, locations and sequencing technologies, and are expressed as Phred Q scores. A Q score of 30 means that the sequencer thinks there is one chance in 1000 that it ‘miss-called’ the base. 30 stands for 10-3. A score of 20 means a 1% estimated chance of being wrong, while 40 means 1 in 10,000. In this field we attach magic importance to reads of Q30 or better, but there is no particular reason for this. Statistics on the quality of reads can be calculated using for example FASTQC, GATK or Antonie (my own tool).

Quality scores over the length of a read
The best reads are typically near Q40, but this still isn’t enough - if we have a 5 million bases long genome and sequence it once, even if all reads were Q40, we’d still end up with 500 errors. If we expect to find maybe one or two actual mutations in our organism, we’d never see them, since they’d be lost in between the 500 “random” errors.
@M01793:3:000000000-A5GLB:1:1101:17450:2079 1:N:0:1
ACCTTCCTTGTTATAGTTTGCGGCCAGCGGTGGCAGTGTCGGCGCTTCTACTAAGGATTCAAGCCCCTGATTTGTGGTTGGATCTGTCNNNNNTACACATCTCCGAGCCCACGAGACAGGCAGAAATCTCGTATGCCGTCTTCTGCTTGA
+
CCDDEFFFFFFFGGGGGGGGGGGGGGHGGGGGGHHGHHHHGGGGGGGGHHHHHHHHHHHHHHHHGHGHGHHHHHHHGHGGGHHHHHHH#####??FFGHHHHHHGGGGGGHGGGGGGHHGGHGGHHHHHHHGGHHHHGGGHGHHHHHHH<
(FASTQ file)
To solve this, we make sure the DNA sequencer doesn’t just read the whole genome once, but maybe 100 times in total. We call this ‘a depth of 100’. Depending on actual error rate, you may need a higher or lower depth. Also, since the machine performs random reads, an average depth of 100 means that there are many areas you only read 10 times (and about a similar amount of areas you needlessly read 190 times!).
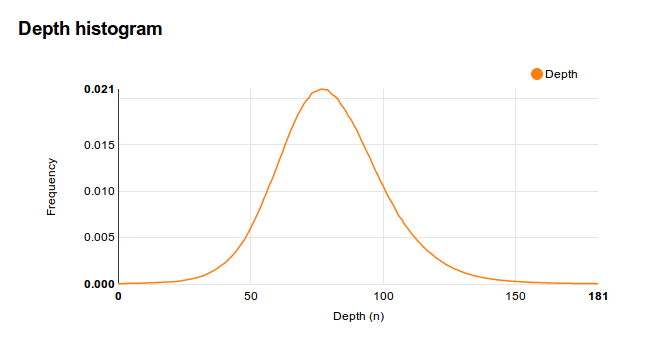
So, we collect a FASTA reference file, prepare our sample, run the DNA sequencer, and get gigabytes of FASTQ file. Now what? Alignment is what. Lots of tools exist for this purpose, famous ones are BWA and Bowtie(2). You can also use my tool Antonie. These index the reference FASTA genome, and ‘map’ the FASTQ to it. They record this mapping (or alignment) in a SAM file.
SAM/BAM Files, Sequence/alignment mapping
This “Sequence Alignment/Map” format records for each ‘read’ to which part of the reference genome it corresponded. Such mapping can be direct, i.e., “it fits here”, but it can also record that a read had extra characters, or conversely, is missing bases with respect to the reference genome. DNA has two strands which are complementary, and are also called the forward and reverse strands. The DNA sequencer doesn’t (can’t) know if it is reading in the backwards or forwards direction, so when mapping, we have to try both directions, and record which one matched.
M01793:3:000000000-A5GLB:1:1101:14433:2944 147 gi|388476123|ref|NC_007779.1| 3600227 42 150M = 3600116 -261 GTCAATTCATTTGAGTTTTAACCTTGCGGCCGTACTCCCCAGGCGGTCGACTTAACGCGTTAGCTCCGGAAGCCACGCCTCAAGGGCACAACCTCCAAGTCGACATCGTTTACGGCGTGGACTACCAGGGTATCTAATCCTGTTTGCTCC
:FHFHFGHHFG0HHHHGHHHFGGCCCFGGFFGFAGGGGGHGGFD?CGGGDHEGGGGGGFHHHEGCGGHHHGEFFG?GHGHHHHHHHGEHG1GHFGFGEFHHHFGHGGHHGFEGGGGHHHHHHHHHGCHGFFGGGGFGGBFFFFF4A@A??
(SAM file)
SAM files are written in ASCII, but are very hard to read for humans. Computers fare a lot better, and tools like Tablet, IGV and samscope can be used to view how the DNA reads map to the reference - where mutations stand out visually. This is important, because some “mutations” may in fact be sequencing artifacts.
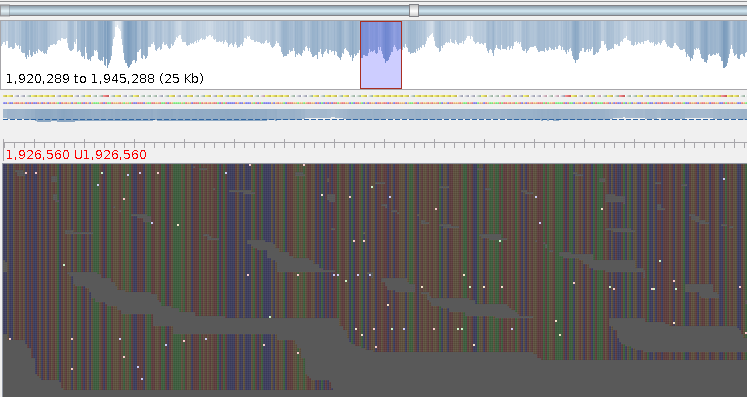
Alignment as viewed in Tablet. White spots are (random) differences between reference & reads
SAM files tend to be very, very large (gigabytes) since they contain the entire contents of the already large FASTQ files, augmented with mapping instructions.
For this reason, SAM files are often converted to their binary equivalent, the BAM file. In addition to being binary, the BAM format is also blockwise compressed using Gzip. If a BAM file is sorted, it can also be indexed, generating a BAM.BAI file. Some tools, like my own Antonie, can emit BAM (and BAI) files directly, but in other scenarios, ‘samtools’ is available to convert from SAM to BAM (and vice versa if required). A more advanced successor to the BAM format is called CRAM, and it achieves even higher compression.
Now that we have the mapping of reads to the reference, we can start looking at the difference between our organism and the reference, and this often leads to the generation of a VCF file, which notes locations of inserts, deletes (together: indels) or different nucleotides (SNPs, pronounced “snips”).
To figure out that these mutations might mean, we can consult a feature database. This is available in various formats, like Genbank or GFF3. GFF3 is best suited for our purposes here, since it precisely complements the FASTA. This General Feature Format file notes for each ‘locus’ on the reference genome if it is part of a gene, and if so, what protein is associated with it. It also notes a lot of other things about regions.
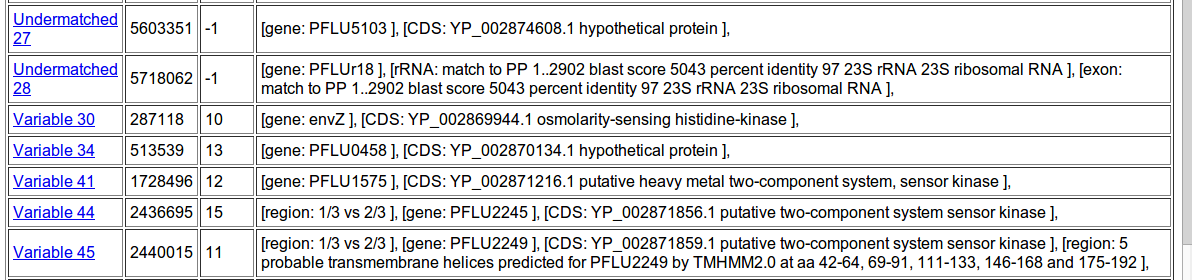
Gene annotations
Summarizing
So, summarizing, a reference genome is stored in a FASTA file, as found on http://ftp.ncbi.nlm.nih.gov/genomes/. A DNA sequencer emits millions of DNA reads in a FASTQ file, which are snippets from random places in the genome. We then ‘map’ these reads to the reference genome, and store this mapping in a large SAM file or a compressed BAM file. Any differences between our organism and the reference can be looked up in Genbank or GFF3 to see what they might mean. Finally, such differences can be stored as a VCF file.
This is the relation between the various file formats:
FASTA + FASTQ -> SAM -> BAM (+ BAM.BAI)
BAM + FASTA -> VCF
But where did the reference come from?
Above, we discussed how to map reads to a reference. But for the vast majority of organisms, no reference is available yet (although most interesting ones have been done already). So how does that work? If a new organism is sequenced, we only have a FASTQ file. However, with sufficient amount of overlap (‘depth’), the reads can be aligned to themselves in a process known as ‘de novo assembly’. This is hard work, and typically involves multiple sequencing technologies.
As an example, many genomes are rife with repetitive regions. It is very difficult to figure out how reads hang together if they are separated by thousands of identical characters! Such projects are typically major publications, and can take years to finish. Furthermore, over time, reference genomes are typically improved - for example, the human genome currently being used as a reference is ‘revision 19’.
Paired end reads
Some sequencing technologies read pieces of DNA from two ends simultaneously. This is possible because DNA actually consists of two parallel strands, and each strand can be read individually. When we do this, we get two ‘paired end reads’ from one stretch of DNA, but we don’t know how long this stretch is. In other words, the two individual reads are ‘close by’ on the chromosome, but we don’t exactly know how close.
Paired end reads are still very useful however, especially since they can disambiguate the correct place because their extra length gives us more context to correctly place them.

Paired end reads are often delivered as two separate FASTQ files, but they can also live together in one file.
Diving in
Next to the wonderful archive of reference materials at http://ftp.ncbi.nlm.nih.gov/genomes/, there are also repositories of actual DNA sequencer output. The situation is a bit confusing in that the NCBI Sequence Read Archive offers by far the best searching experience, but the European Nucleotide Archive contains the same data in a more directly useful format. The Sequence Read Archive stores reads in the SRA format, which requires slow tools to convert to FASTQ. The ENA however stores simple Gzip compressed FASTQ, but offers a less powerful web interface. It might be best to search for data in the SRA and then download it from the ENA!
Introducing Antonie
So when I entered this field, I had a hard time learning about all the file formats and what tools to use. It also didn’t help that I started out with bad data that was nevertheless accepted without comment by the widely used programs I found - except that meaningless results came out. Any bioinformatician worth her salt would’ve spotted the issue quickly enough, but this had me worried over the process.
If desired answers come out of a sequencing run, everyone will plot their graphs and publish their papers. But if undesired or unexpected answers come out, people will reach for tooling to figure out what went wrong. Possibly they’ll find ways to mend their data, possibly they’ll file their data and not publish. The net effect is a publication bias towards publishing some kind of result - whether the origin is physical/biological, or a preparation error. In effect, this is bad science.
From this I resolved to write software that would ALWAYS analyse the data in all ways possible, and simply refuse to continue if the input made no sense.
Secondly, I worried about the large number of steps involved in typical DNA analysis. Although tools like Galaxy can create reproducible pipelines, determining the provenance of results with a complicated pipeline is hard. Dedicated bioinformaticians will be able to do it. Biologists and medical researchers under publication pressure may not have the time (or skills).
From this I resolved to make Antonie as much of a one-step process as possible. This means that without further conversions, Antonie can go from (compressed) FASTQ straight to an annotated and indexed BAM file with verbose statistics, all in one step. Such analysis would normally require invoking BWA/Bowtie, FASTQC, GATK, samtools view, samtools sort, samtools index & samtools mpileup. I don’t even know what tool would annotate the VCF file with GFF3 data.
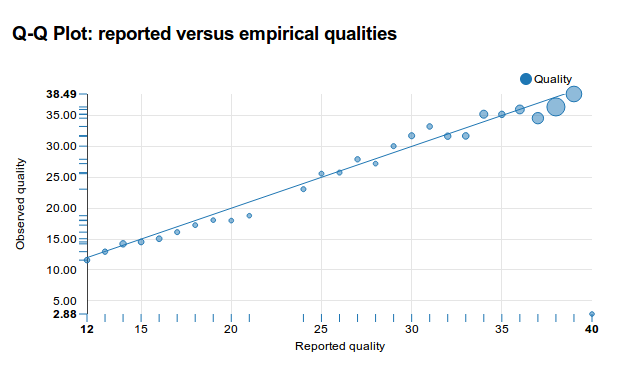
“Well-calibrated measurements”
Finally, with decreasing sequencing costs (you can do a very credible whole genome sequencing run of a typical bacterium for 250 euros of consumables these days), the relative costs of analysis are skyrocketing. Put another way, 10,000 euros would a few years ago net you one sequencing run plus analysis. These days (2013), the same amount of money could net you 40 sequencing runs but no analysis, as outlined in “The $1000 genome, the $100000 analysis?”.
Because of this, I think the field should move to being able to operate with (minimal) bioinformatician assistance for common tasks. End-users should be able to confidently run their own analyses, and get robust results. Software should have enough safeguards to spot dodgy or insufficient data, software should be hard to misuse.
Antonie isn’t there yet, but I’m aiming towards making myself redundant at the lab (or conversely, available for more complicated or newer things).
>For reasons I don’t understand, the FASTA file(s) have a .fna file extension.
ReplyDeletefna = nucleic acid, faa = amino acid. I've also seen fasta and fa.
Can Antonie deal with RNA-Seq data?
Hi DJ, thanks! Yes, we've processed RNA-Seq with Antonie, but we don't do junctions etc, but the data we got was useful enough. I think it would fit in the profile to fully automate this stuff too.
ReplyDeleteSo when is the time antonie or similar devices can be operated by dummies Bert?
ReplyDeletenot that hard to do today. It is the 100k$ MiSeq with the $1000 reagent kit that hurts most ;-)
DeleteThis comment has been removed by the author.
ReplyDeleteOnly small data?
ReplyDeleteCan I use it to analyse human sequence data?